FDA

In this week’s Digital Health Roundup, Medtech Insight’s Ryan Nelson highlights Click Therapeutics’ FDA-cleared digital therapeutics (DTx) for depression and Sinaptica Therapeutics’ personalized neuromodulation for Alzheimer’s patients. Marion Webb discusses her interview with MindMaze’s John Krakauer on their gaming-focused DTx to help people recover from serious brain injuries. Elizabeth Orr introduces new voting members of the new Digital Health Advisory Committee and Natasha Barrow discusses Hello Heart’s new symptom-tracking feature in their heart-focused app.
In this week’s Digital Health Roundup, Medtech Insight’s Ryan Nelson highlights Click Therapeutics’ FDA-cleared digital therapeutics (DTx) for depression and Sinaptica Therapeutics’ personalized neuromodulation for Alzheimer’s patients. Marion Webb discusses her interview with MindMaze’s John Krakauer on their gaming-focused DTx to help people recover from serious brain injuries. Elizabeth Orr introduces new voting members of the new Digital Health Advisory Committee and Natasha Barrow discusses Hello Heart’s new symptom-tracking feature in their heart-focused app.

A draft guidance document opened for comment by the FDA offers additional Q&As on issues around biosimilar applications and interchangeability.

Richard Lowenthal, co-founder, and CEO of ARS Pharma, highlights the crucial unmet need for needle-free injections. Challenging issues posed by current epinephrine injectors, In Vivo questions the current and future progression of Neffy.

Brazil has also published its recent regulatory reliance regulations in English, a move designed to increase confidence in its decisions as it presses on with aligning its national practices with global best practices.

This week, Neuralink announced it received US FDA breakthrough device designation for a device to restore sight; medtechs Discure and DeepLook secured new funding; FDA pump recalls from B. Braun Medical and Fresenius Kabi; Axonics prevails in patent infringement lawsuit with Medtronic; Merit Medical buys Cook Medical for $210m.

This week, two device testing labs in China landed FDA warning letters; refunds for 1Health.io clients; FDA AR/VR product list expands.

The Food and Drug Administration’s final guidance outlines the agency’s thinking for sponsors submitting de novo requests electronically, which they must starting in October 2025.

Proposal to have panelists rate the strength of efficacy and safety evidence on a numeric scale also draws support in written comments on ways to optimize the advisory committee process.
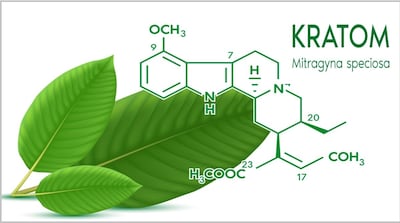
Week after publishing safety alert about OPMS Black Liquid Kratom “linked to serious adverse health effects, including death,” FDA announced market research “to understand and characterize emergent risk/safety and perceived benefits reportedly linked to kratom and psychedelics.” But it withdrew the study 10 days later.

Semi-annual regulatory agenda of non-binding target dates also sets December goal for an NPRM to recognize N-acetyl-L-cysteine as a lawful dietary ingredient. Like IND exemptions rule, item on NAC included for first time in FDA’s list.

This week, a medical group sued the FDA to block a lab-developed test rule; the FDA published guidance on device classifications; Defibtec issued a recall of its chest compression device and ICU Medical updated instructions for its infusion pump batteries; Maui Imaging raised a $4m DOD grant to put imaging tech into military-based trauma units.

Conflicted experts should be allowed to participate as nonvoting members and panels should take a benefit-risk vote on product-specific applications, the majority of respondents said in a survey conducted by 3D Communications.

But 2024 will see a lull between two strong years.

The US FDA has published draft guidance for predetermined change control plans for medical devices along with recommendations for sponsors including them in marketing submissions to the agency.

Linvoseltamab has missed out on an August US approval, but Regeneron remains confident it will emerge as the best-in-class BCMAxCD3 bispecific therapy.

Former FDA device center head Jeffrey Shuren is facing conflict of interest claims for not recusing himself from regulatory decisions involving clients of his wife’s legal firm. Critics call for an investigation and stricter ethics compliance.

Pink Sheet infographic shows that while overall growth continues at the drugs and biologics centers, the US FDA still must add many employees to meet user fee-mandated hiring goals.

Letter to US FDA by a bipartisan group of House members cites clinical trials sponsored by Eli Lilly and Pfizer as examples of a widespread industry practice. But any legislation to curtail such practices is not expected to be part of the BIOSECURE Act.

A government report requested by US lawmakers Debbie Dingell and Anna Eshoo to review the FDA’s postmarket surveillance of medical devices stresses that strengthening the system is critical to addressing adverse events linked to devices after they hit the market.
ADVERTISEMENT