Quality Control

This week, Nipro Medical Corp. announced it will invest $397.8m to build a US-based production plant, generating 232 new jobs; both Baxter and Hamilton announced ventilator recalls; Imperative Care wins FDA clearance for its stroke catheter; Intelligent Ultrasound Group plc entered into a conditional sale and purchase deal to sell its Clinical AI business to GE HealthCare for £40.5m; RMI distributed 350m rapid test kits in the fight against HIV/AIDS; Jiangsu Shenli Medical Production Co., Ltd received a second FDA warning letter about quality and safety of plastic syringes.

Cutting edge technology could significantly enhance operating room efficiency, according to a new report from Medtronic. The problem, however, is that most ORs don’t have it.

A new round of tariffs imposed by the Biden administration on various Chinese goods, including medical devices, points to a broader shift in US strategy for strengthening supply chains and ensuring Americans have reliable access to safe products, according to analysts who spoke to Medtech Insight about the tariffs. While fueled by the pandemic, the momentum pushing this change in trade policy has been growing for some time.

The US FDA has issued a final rule allowing the agency to destroy some medical devices that have been refused entry into the US. The rule takes effect 1 July.

The US FDA has issued a final rule allowing the agency to destroy some medical devices that have been refused entry into the US. The rule takes effect 1 July.

This week, Philips Respironics reached a $1.1b settlement affecting CPAP and other breathing devices. Toku announced it received US FDA breakthrough device designation for its MyKidneyAI technology. This May, the FDA will hold its REdI conference focusing on innovation in medical product development and hold another townhall focusing on considerations for selecting a sterilization modality.

Translating research from proof of concept to clinical investigations is a difficult hurdle to overcome. To succeed, researchers need to design their technology for industrial standard manufacturing early on, Anne Vanhoestenberghe, director for the Manufacture of Active Implants and Surgical Instruments (MAISI), told Medtech Insight.

The US FDA’s device center has unveiled a new public dataset designed to assist chemistry laboratories in ensuring the robustness of chemical characterization methods used to assess the biocompatibility of medical devices.

A consent decree agreed to in January between Royal Philips and the US government is now official. The decree stems from a recall in 2021 of millions of the company’s sleep therapy and breathing devices due to risks posed by the sound abatement foam inside the machines.

BD CTO Beth McCombs discusses her role in driving innovation, including the integration of AI and generative AI to create efficiencies, and passion for creating a culture of inclusion, diversity and equity.

The US FDA has completed the final rule that will harmonize its decades-old quality system regulations with international standards. The new Quality Management System Regulation aims to reduce the regulatory burden on device makers by linking domestic and international requirements.

The FDA has revised draft guidance issued in 2022 on how the agency plans to continue using remote technology to assess regulated facilities. The FDA established remote assessments in early 2021 to keep up its regulatory obligations during the COVID-19 pandemic.

The US FDA issued a stern warning to Fresenius Medical Care over its slow approach to corrective actions over potentially harmful toxins emitted from its hemodialysis machines and accessories.

A new annual report argues that last year was both transformative and historic for the US FDA’s Center for Devices and Radiological Health. The center authorized the highest number of novel medical devices in history while turning the corner on the pandemic.
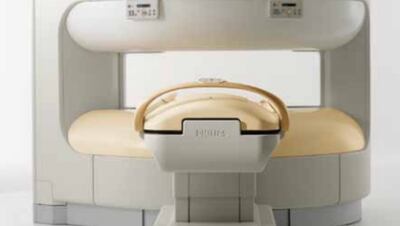
Philips initiated a recall in November of its Panorama 1.0T magnetic resonance system due to the risk the system could explode during use. The FDA has designated the recall class I.

As the US FDA works to finalize new regulation of lab-developed tests, it must consider more than two thousand comments that have poured into the agency since the proposed rule was published in October. The comment period closes Monday.

The US FDA has received a significant increase in medical device reports associated with Philips DreamStation 2 machines, including fire, smoke, and burns.

Three years after joining, the US FDA announced its withdrawal from the Global Harmonization Working Party and intention to focus its work on medical device harmonization through its collaboration with the International Medical Device Regulators Forum.

The US FDA has published final guidance requiring firms to notify the agency about significant manufacturing interruptions of medical devices during a public health emergency, as well as a supplementary draft guidance that includes a list of specific devices that manufacturers must report to the agency when they are in short supply.

During a recent webinar, Elizabeth Hillebrenner of the US FDA’s device center sought to clarify specifics on the agency’s proposed rule to regulate laboratory-developed tests, which establishes FDA oversight of the tests over a five-year period.
ADVERTISEMENT